


Hit the Vintage Store and Get Yourself Some CHEAP PLAID
Compressed Hourglass Embedding Adaptations of Proteins & Protein Latent Induced Diffusion: Two-stage latent diffusion for all-atom protein co-generation


The machine learning for protein design community is obsessed with co-generating sequence & structure of proteins simultaneously. Why? Mostly because images of protein structure like this:

are cooler than protein sequences like this:
GYDPETGTWGand are more fun to look at.
OK there are scientific reasons too, summarized very nicely in this review article (emphasis mine):
Structure-based methods leverage more semantically rich information and are most effective when the desired function is mediated by a small structural motif, but structural data is limited and biased, leading to less diverse learned distributions. Meanwhile, sequence-based methods generalize more easily to functions that rely on larger or multiple domains, are mediated by a structural ensemble, or involve intrinsically-disordered regions…To overcome the limitations of generating sequence or structure alone, researchers are developing sequence structure co-generation methods, which aim to model and generate both modalities simultaneously. Reasoning about sequence and structure throughout generation and fully leveraging the available data should result in a more accurate learned distribution and more plausible generations.
There are two key points here. 1) There are settings where well-defined structural motifs mediate well-understood interactions and functions. In those cases, having all-atom structures allows us to evaluate and reason about our protein design methods in ways that are difficult or impossible when reasoning over sequence alone. And 2) There is information content in the hundreds of thousands of crystal structures contained in the PDB, and we should take advantage of it.
So how to do it? How do we generate sequence and structure simultaneously? Well, if we’re going to be serious about it, we better generate all-atom structures (backbones and side chains), so we can really evaluate everything that protein structure gives us access to. We better include big sequence datasets too, since one of our goals is to learn from all the data.
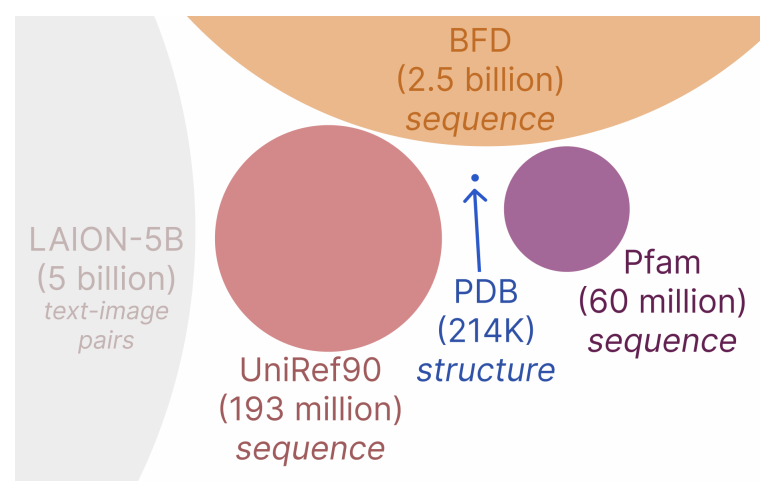
The big idea: repurposing a biological foundation model
A brilliant idea 💡 that feels like it should work is to repurpose a biological foundation model that has already been trained on sequence and structure data: ESMFold. ESMFold is trained to predict all-atom structures from single sequence inputs. If we could sample in the latent space of that model, we could decode out new sequences and all-atom structures. We could train a diffusion model on the latent space and learn to sample from it. Let’s try it!
It turns out that the language model part of ESMFold, ESM2-3B, has massive activations 🥴. That means the latent space is pathologically disorganized, where some activations in the model are 3000x larger than others. Massive activations have been observed in large language models (LLMs) as well. They affect <0.01% of values, but that’s enough to really mess things up. That makes it impossible to learn a diffusion over this space and enable co-generation.1

Luckily, once the problem is diagnosed, there are some clear options: 1) Spend a few million $$ to train a new ESMFold that doesn’t have this problem, or 2) perform surgery on the model and remove the massive activations. Unfortunately, (1) will have to wait for a philanthropic cash infusion, and (2) doesn’t work because the model actually relies quite heavily on these massive activations and removing them destroys the performance. OK, we need better options.
Luckily, there’s a CHEAPer alternative. CHEAP = Compressed Hourglass Embedding Adaptations of Proteins. First, we apply a per-channel normalization to tame the massive activations, and then we introduce a compression scheme that forms a new, smaller, smoother latent space. Inspired by two-stage image diffusion models, we learn a diffusion over this new, compressed space, do our sampling there, and then upsample back to real sequences and structures. It’s like learning to generate small, lower resolution images and then refining them to create bigger, high-res images.
We can actually compress the original embeddings a lot, by a factor of 32x, and still reconstruct the all-atom structure to within measurement error (~1Å). We want to know how much “information” is contained in the sequence-structure embedding, rather than assume that the structure information is valuable, so we also examined how well our compressed embeddings do when we use them to predict protein properties or function. Interestingly, some properties (solubility, binary localization, stability) are very easy to predict from highly compressed embeddings, while others (fluorescence and subcellular localization) are more sensitive to information loss after aggressive compression.
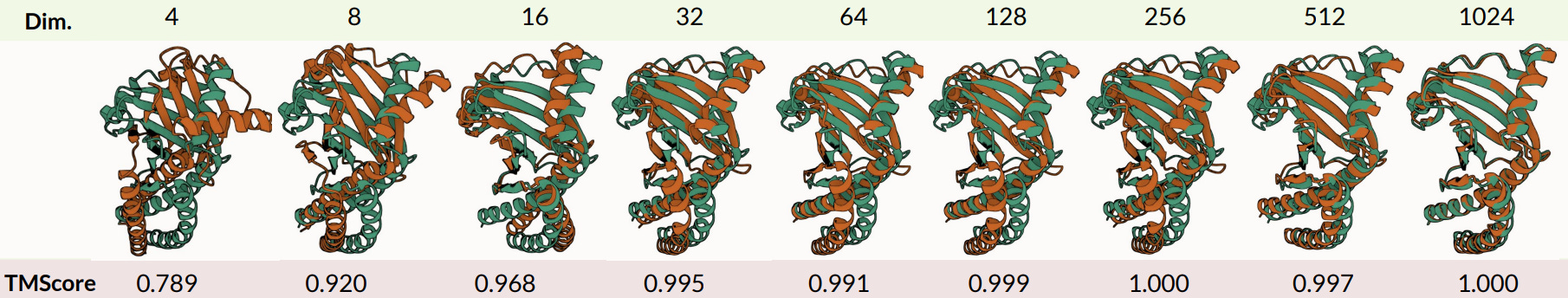
Tamed latent space → latent diffusion
Now we’re ready to tackle our original goal of enabling all-atom co-generation, while leveraging all available sequence and structure data. PLAID = Protein Latent Induced Diffusion. PLAID uses a big diffusion transformer (DiT) to learn a diffusion over the compressed CHEAP embedding space, which we can efficiently sample from and decode back to sequences & all-atom structures. And…it works!

PLAID can “access” all protein space across sequence and structure databases, and it generates highly diverse, high quality (high pLDDT) structures. Because of our compression scheme and sequence database training, PLAID can also generate longer, bigger proteins than other methods.
We’ve trained on all the data, we’re getting beautiful structures, now what new things can we do with this model?
To get at the heart of what makes co-generation methods unique and useful, we also equipped PLAID with function conditioning. We can control PLAID generation via motif scaffolding (specifying part of a structure to preserve during sampling), and by conditioning on species and function. This is what we’re after, because now we can include prompts like “Human” and “Transmembrane transporter activity” or “Mouse” and “heme binding” and look for conserved motifs at active sites that give us confidence we’re generating proteins that might actually have the desired functionality. Because we’re generating side chains as well, we can look at atomistic detail of these active sites and make sure our generated designs are biophysically reasonable compared to what’s been observed in humans and animals.
Where do we go from here (some free research ideas)?
A free research idea that is not too expensive to do is to test PLAID in the lab! If you are interested in motif scaffolding and/or function-guided design and think an all-atom co-generation model would be a cool way to do it, we’ve got the method for you.
Likewise, if you’ve been focused on latent diffusion for co-generation, or all-atom co-generation, because you felt like it should work, it does! Whatever applications or ideas you were dreaming of, you can try them out with PLAID.
There are also plenty of opportunities to improve upon PLAID. You could try non-diffusion sampling schemes, like walk jump or flow matching. If you have GPU hours to spare, you might also like to develop a successor to ESMFold (PLAIDFold? CHEAPFold?), making the improvements we identified, and PLAID would certainly work even better.
Resources and contact
Acknowledgements
CHEAP and PLAID were entirely led by Amy X. Lu, and all the credit for getting this stuff to work and doing amazing research goes to her. Any time I (Nathan) wrote “we” in this post, that really means “Amy + her supporters.” Thanks to all our collaborators and co-authors on this research.
 | A guest post by |