
Reading Between the Isoforms: Interpretable Machine Learning Meets Long-Read RNA-Seq for Biomarker Discovery
A guest post by Isabelle Heifetz Ament


Isabelle is a PhD student at my alma mater, the University of Pennsylvania, in the Lin Lab at the Children’s Hospital of Philadelphia (CHOP). The Lin Lab developed TEQUILA-seq, a low-cost method for long-read RNA sequencing. Isabelle’s PhD research involves using long-read RNA sequencing for exploring transcriptome variations in human diseases. Isabelle and I met as part of the Penn PhD Career Exploration Fellowship (CEF), where I was fortunate enough to first get exposure to the biopharma industry many years ago through a CEF with Merck. When I told Isabelle that I knew nothing about long-read RNA-seq, but that I’d be excited to learn from her and explore how modern computational approaches could help in her research, Isabelle fearlessly jumped into the world of AI and ML. This post is a result of those investigations. I believe the future of biology research belongs to scientists like Isabelle, who know what the important biological questions are to ask, and use AI tools to explore previously unreachable areas of biology. – Nathan

Two decades ago, sequencing a human genome was a once-in-a-generation achievement. The first drafts, published in 2001, took over a decade of coordinated effort, billions of dollars, and cutting-edge technology. Today, the same feat can be done in days for a few hundred dollars.
Sequencing is no longer limited to bulk DNA. Modern platforms can profile the complete RNA content of thousands of individual cells, map gene expression in intact tissues, and even recover genetic material from centuries-old specimens. Targeted approaches now enable rapid genetic diagnoses, while single-cell and spatial methods give unprecedented views of cell identity and tissue organization.
Despite these advances, the most widely used transcriptomic method – short-read RNA sequencing (RNA-seq) – has inherent blind spots. This approach fragments RNA into 100–300 base pair pieces before sequencing and computationally reconstructing transcripts. While it produces high-quality data for well-annotated genes, it struggles to capture full-length isoforms, complex splicing patterns, and sequences from repetitive or poorly annotated regions. Most pipelines filter out reads mapping to multiple loci, resulting in reduced read coverage for repetitive loci like long noncoding RNAs (lncRNAs) and transposable elements (TEs), which together make up more than half of the human genome.
Machine learning (ML) has become a powerful tool for transcriptomic analysis, particularly in single-cell RNA sequencing (scRNA-seq). With short-read data, ML frameworks can classify cell types, infer developmental trajectories, and even model perturbation responses in silico. More recently, ML-driven integration of spatial sequencing and scRNA-seq data has also enabled sensitive detection of changes in cellular composition and tissue architecture across time points or between conditions. ML has even been applied to predict patient response to treatments using patient scRNA-seq input. Together, ML and short-read RNA-seq show great promise for transforming our ability to dissect cell identity and transcriptional dynamics in health and disease.
Yet these gains are constrained by the limitations of short-read sequencing: current models often rely on gene-level summaries, missing isoform diversity and regulatory features embedded in complex or repetitive regions. Long-read RNA-seq overcomes these barriers by sequencing transcripts end-to-end, revealing complete isoform structures, novel splice junctions, TE exonization events, and unannotated transcripts from the so-called “dark genome.” This isoform-resolved view opens new opportunities for ML models to learn from the complete architecture of the transcriptome.
The convergence of long-read sequencing and interpretable ML sets the stage for a new frontier: identifying biologically meaningful isoform variation across disease and cell identity contexts. In this review, we outline how combining these tools can enhance sensitivity, resolution, and biological relevance in transcriptomic analysis. We focus on key frameworks, including scVI, VEGA, devCellPy, scGPT, and XGBoost + SHAP, and discuss how they can be adapted to leverage isoform-level features, paving the way toward biomarker discovery in regions of the genome that have traditionally been overlooked. Unlocking this “dark genome” space could reveal entire classes of clinically relevant biomarkers that have remained invisible to conventional analyses — enabling earlier detection, improved stratification, and new therapeutic avenues.
The Challenge:
Roughly half of the human genome is repetitive, much of it derived from ancient viral insertions known as TEs. Far from being inert “junk,” recent data supports that TEs remain transcriptionally active or have been co-opted into gene regulation. They can contribute alternative promoters, splice sites, and coding or noncoding exons. Alongside novel lncRNAs and alternative splicing, these elements add another layer of complexity to the transcriptome, particularly in development, cancer, and immune activation.
If we want to identify TE-derived biomarkers or cell-type–specific, alternatively-spliced isoforms that are relevant to health and disease, short-read RNA-seq falls short.
Long-read RNA-seq solves many of these problems. By sequencing transcripts from end to end, it preserves isoform structures, resolves usage patterns, and opens access to previously unmappable regions. This means we can finally see the diversity of TE-derived and novel transcripts in full detail. But it also creates a new problem: instead of a few hundred annotated isoforms, we now have thousands (many lowly expressed, poorly annotated, or unique to specific conditions or cell types.)
To pinpoint the features that truly distinguish cell types or disease states, we need more than traditional differential expression tests. We need models that can learn from transcript-level features, find predictive patterns across them, and offer results that are biologically interpretable.
From Long Reads to Features That Matter
Long-read sequencing gives us the transcripts; feature engineering makes them usable for ML. Each transcript can be represented as a vector of interpretable features, such as:
Expression: read counts, counts per million (CPM), isoform ratios
Structure: Number of exons, splice junction patterns, retained introns
Annotation: Whether it's known, novel, TE-overlapping, lncRNA-associated
Coding potential: Predicted Open Reading Frame (ORF) presence, peptide hits
Regulatory context: Methylation, conservation, chromatin state
These features form a matrix where each row is a transcript and each column is a property that might help us understand biology. And now, we can model.
The Models:
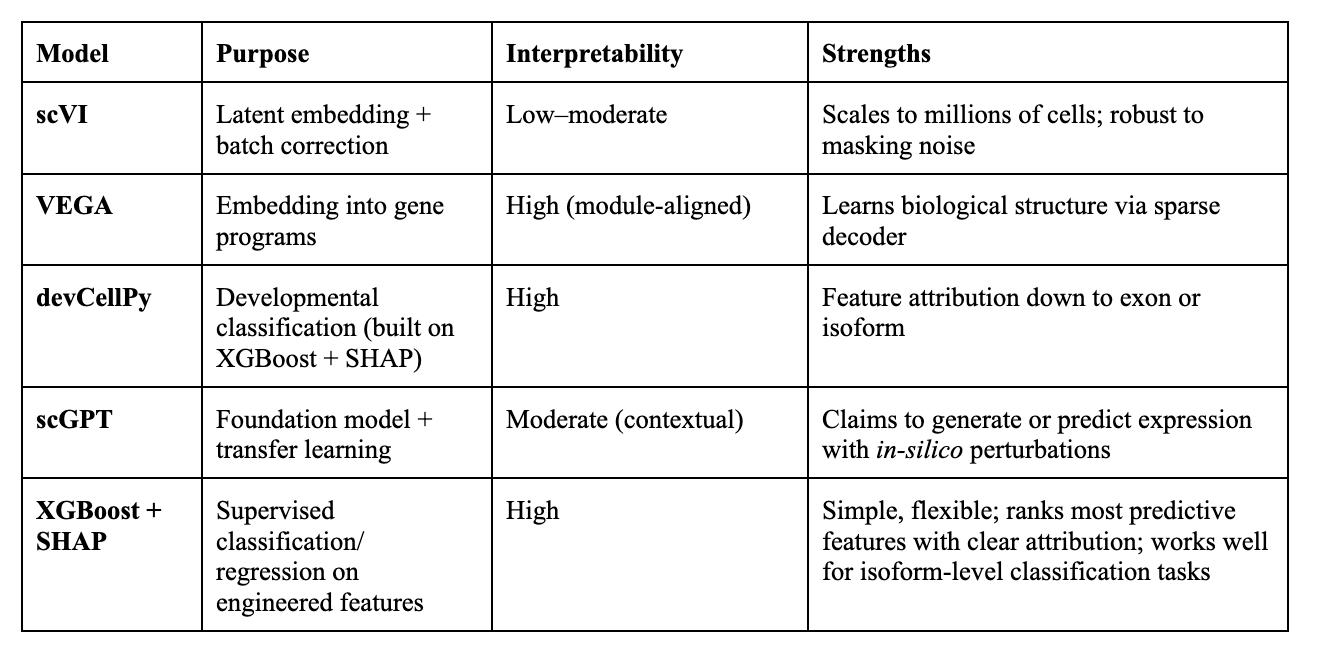
Interpretability:
High : Directly links outputs to specific biological features (genes, isoforms, exons)
Moderate : Provides clues (attention weights, correlations) but needs extra steps to connect to biology
Low : Little to no direct mapping to biological features without heavy post-hoc analysis
Why Not Just Do Differential Expression?
For many biologists, differential gene expression (DGE) analysis is the starting point for turning sequencing data into hypotheses. It’s a trusted workhorse for identifying transcripts that change in abundance between conditions. But with isoform-level data from long-read sequencing, DGE shows its limits.
ML can address many of these gaps by learning from multiple transcript features at once and — if interpretable — explaining which features drive the predictions. The key differences are:
DGE tests transcripts in isolation
DGE asks whether a single transcript is consistently up or downregulated. It misses combinations that are predictive together.
ML can learn that a specific pair of isoforms – for example, a TE-derived exon plus a splicing change – distinguishes two conditions even if neither is significant alone.
DGE assumes linear changes
DGE is tuned for smooth, proportional shifts. It can miss threshold effects, where a transcript only matters above (or below) a certain level.
ML can model such nonlinear patterns directly.
DGE struggles with noisy, repetitive features
Transcripts from repetitive regions are often lowly expressed and variable. DGE may filter them out or fail to detect significance.
ML can combine multiple weak signals into a strong predictor and rank their importance.
ML adds interpretability for discovery
Interpretable ML frameworks (e.g., XGBoost + SHAP, devCellPy) produce importance scores for each feature, revealing which isoforms, splicing events, or structural elements contribute most to a classification.
Example:
Imagine a study of brain tumors using long-read RNA-seq. Two novel isoforms are detected:
Isoform A: contains an exon derived from a TE
Isoform B: has a retained intron in a neural lncRNA
Both are rare and variable in expression. Because one overlaps a repetitive element, and the other is lowly expressed, each would likely be filtered out or fail to reach significance in a standard DGE analysis.
A supervised ML model, trained to distinguish tumor from normal samples, instead learns that the combined expression of these two isoforms is highly predictive of tumor status. Using an interpretable approach (e.g., XGBoost + SHAP), the model ranks them among the top features.
This example illustrates why ML — paired with long-read sequencing — is essential for finding predictive isoform signatures that are multivariate, nonlinear, and often hidden in repetitive or poorly annotated regions of the genome.
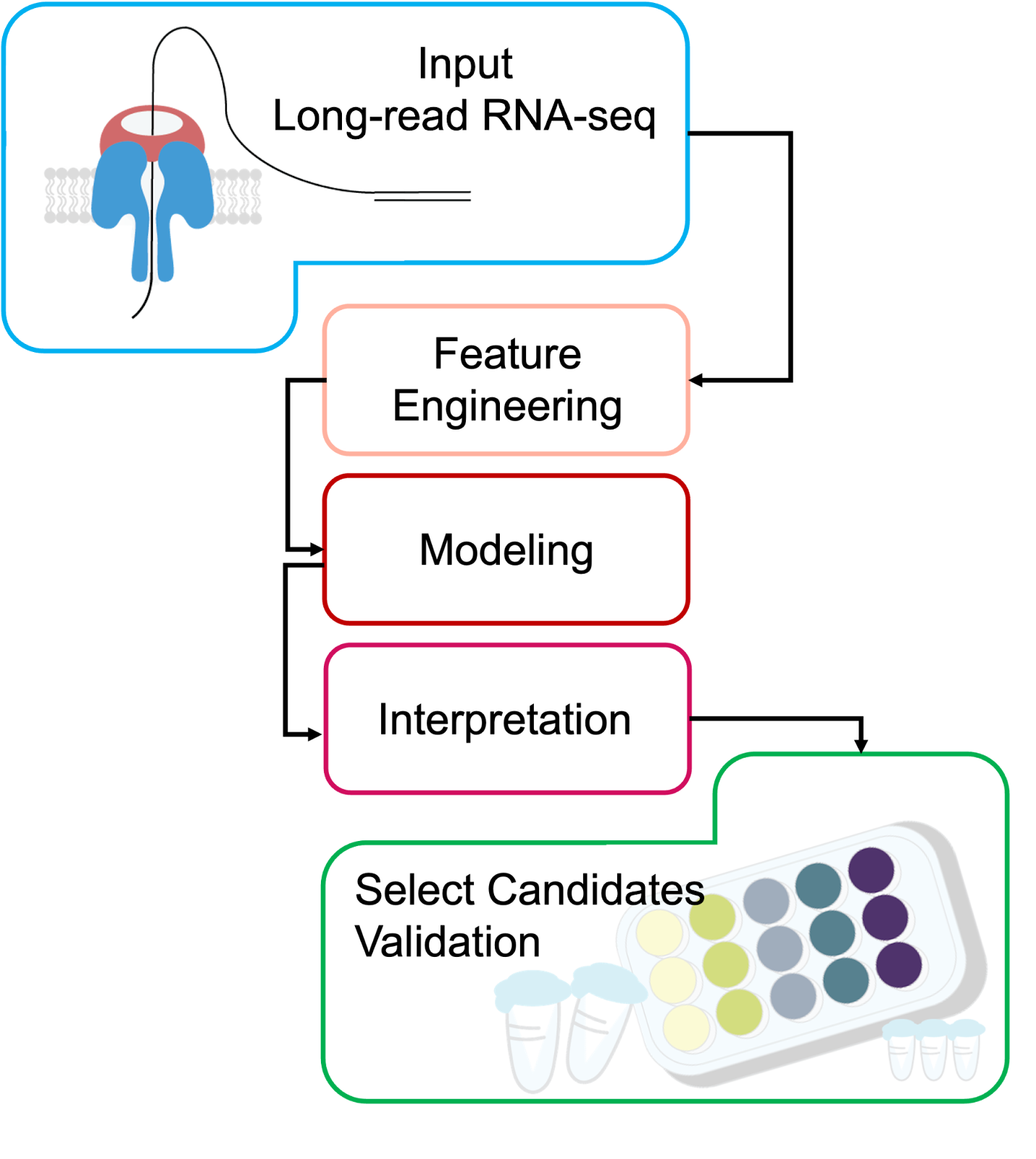
A Blueprint for Discovery
The same logic from the brain tumor example can be generalized into a systematic pipeline for discovering isoform- or TE-based biomarkers from long-read RNA-seq:
Input
Collect long-read RNA-seq data from relevant samples
Optional: integrate matched single-cell or spatial transcriptomics to pinpoint cell-type or microenvironment-specific expression
Feature Engineering (See Above)
Modeling
Noise reduction / structure exploration: scVI or VEGA to capture latent structure while preserving interpretability.
Classification and ranking: XGBoost or devCellPy to predict condition labels and identify top-ranked features.
Simulation / transfer learning: scGPT to model perturbations or integrate across datasets.
Interpretation
Use SHAP values (tree-based models) or decoder weights (VAEs) to highlight the isoforms, splicing events, or TE-derived transcripts most predictive of the condition.
Validation
Molecular confirmation (e.g., RT-PCR and sequencing, qRT-PCR, targeted long-read amplicon sequencing) to verify isoform structure and abundance.
Functional assays – perturbation experiments (e.g., overexpression, knockdown, CRISPR editing), reporter assays for regulatory elements, or protein-level detection.
Together, these experiments verify that the predicted transcript exists in the cell and plays a role in the phenotype of interest, providing confidence that the pipeline is producing biologically meaningful outputs.
In the brain tumor scenario, this pipeline would identify Isoform A and Isoform B as high-ranking features, validate their tumor-specific expression, and assess whether either is presented as an antigen or functionally drives disease.
Frontiers: From Biomarker to Therapeutic
When combined with single-cell or spatial data, this workflow can reveal cell-type–specific, alternatively-spliced and/or TE-derived transcripts that define unique microenvironments in disease. In oncology, such transcripts could serve as:
Targets — if presented as tumor-specific antigens for immunotherapy.
Triggers — in approaches that deliberately induce TE expression to provoke immune recognition.
Beyond cancer, similar strategies could uncover isoform signatures in autoimmunity, neurodegeneration, or infectious disease — conditions where repetitive or poorly annotated transcripts are often ignored in standard analyses.
Limitations and Next Steps
Long-read RNA-seq, paired with machine learning, has the potential to uncover biomarkers hidden in poorly annotated, repetitive, and low-abundance regions of the genome—capturing isoform-level splicing, lncRNA expression, and transposable element activity with locus-level precision. To fully realize this promise, the field must prioritize three things:
Sequencing with fewer errors to improve isoform and ORF calls
Richer and more complete reference annotations for novel genes and elements
Large, diverse, well-phenotyped long-read datasets with standardized benchmarks.
With more confident datasets and stronger annotations, current ML frameworks will be able to make reliable, biologically meaningful predictions across the full complexity of the transcriptome. The result could be truly transformative, unlocking new layers of biology and accelerating the discovery of meaningful, actionable biomarkers for human health.
Getting in touch
If you liked this post or have any questions, feel free to reach out to Isabelle over email (iament@sas.upenn.edu). You can email or connect with Nathan on LinkedIn and X / Twitter.
 | A guest post by
|